Microbial diversity research
Life on Earth has evolved for millions of years, creating a vast diversity. Even though it's not apparent to us, most of this diversity is microbial. Exploring all of these microbes out there is challenging, though. They are tiny and often morphologically hardly distinguishable. Luckily, over the last ten years, molecular techniques such as high throughput sequencing have become more available. These modern methods allowed microbial diversity research to scale. One of the popular high throughput sequencing methods is environmental sequencing. Environmental sequencing enables us to sequence numerous organisms within the same sample. We don't need to painstakingly isolate all the individual specimens one by one but sequence the whole sample from the environment.
Environmental sequencing and phylogenetic placement
Environmental sequencing yields a large number of sequences that we need to match with the correct taxonomic names. There are several ways how to do it. One of them is placing the environmental sequences on the phylogenetic tree that is made from already identified taxa. So we can identify the phylogenetic position of environmental sequences. This method is called phylogenetic placement. The tree where we place the sequences is called a reference tree.
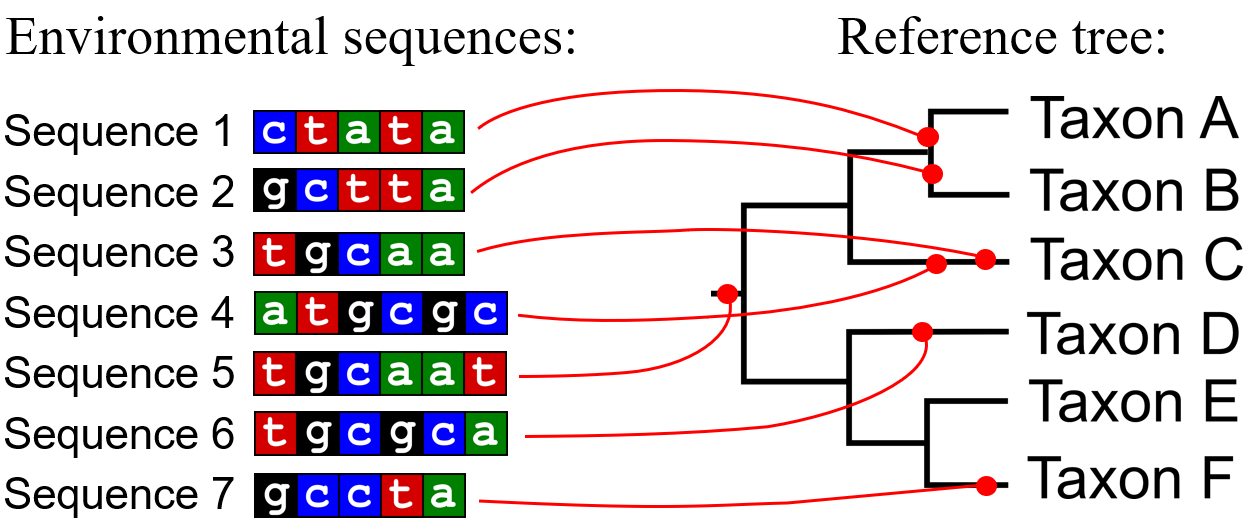
Figure 1. Phylogenetic placement.
A reference tree could be very broad, containing all three main domains of life. Or we can focus on a specific group, for instance, a tree that would include only ciliates, so in that case, we can analyze ciliate diversity in more depth.
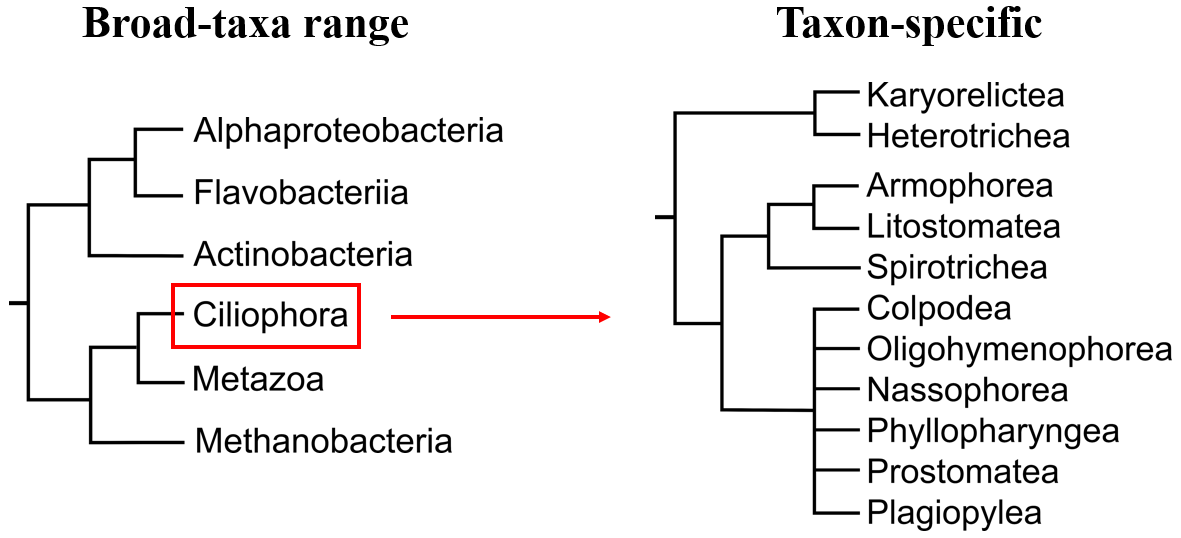
Figure 2. Reference tree examples.
The problem is that reference trees are missing for many groups or are not publicly available. On the other hand, environmental molecular data are still increasing, so many specialists can benefit from this data. They can assess the diversity of the group of their interest by phylogenetic placement. One of the examples are ciliates. Although each global diversity survey shows ciliates as one of the most dominant eukaryotic single-cell groups, there are only a few ciliate-specific molecular diversity studies. Therefore, we decided to fill this gap and design a global ciliate SSU-rRNA tree that may be used in future studies. We also wanted to make 1) several versions of the reference trees, each version suitable for a different sequence platform, and 2) choose the most practical and followed taxonomy.
Our reference trees
The very first step was creating a dataset with raw ciliate sequences. We took advantage of the already existing ciliate dataset from 2014, and we enriched this dataset with since-published ciliate sequences. But we included only morphologically-characterized sequences from GenBank. Environmental sequences were not included except cariacothrixs because they have only environmental data. We prioritized sequences from the recent ciliate phylogenetic studies to include not just taxonomical diversity (e.g., to have every genus represented) but also phylogenetic diversity (e.g., include every well-supported distinct clade). We ended up with 478 ciliate and six outgroup taxa. This dataset contains the most ciliate molecular diversity but is still not too overwhelmingly big. The tree from this dataset is possible to visualize and work with easily.
In the next step, we aligned all sequences from the dataset. We created three different alignments. Each alignment has different masking, which means that the uncertainly aligned positions were in each alignment treated differently. In the first alignment, we kept all nucleotide positions, even those that were very ambiguously aligned. This alignment is the most phylogenetically informative and could be a good option for long reads from sequencing platforms such as PacBio. In the second alignment, all ambiguously aligned positions were masked. Therefore, this alignment is more conservative and may be a good option for studies focused on the higher taxon levels. In the third alignment, ambiguously aligned positions were also masked, except for the V4 region. The V4 region is often a target part when short-read sequencing platforms are used, such as Illumina. This alignment is a good option if you have environmental sequences from the V4 region, which is often the case. In total, we created three alignments, each for a different purpose and sequencing strategy.

Figure 3. Three different alignments that we designed.
Taxonomy used
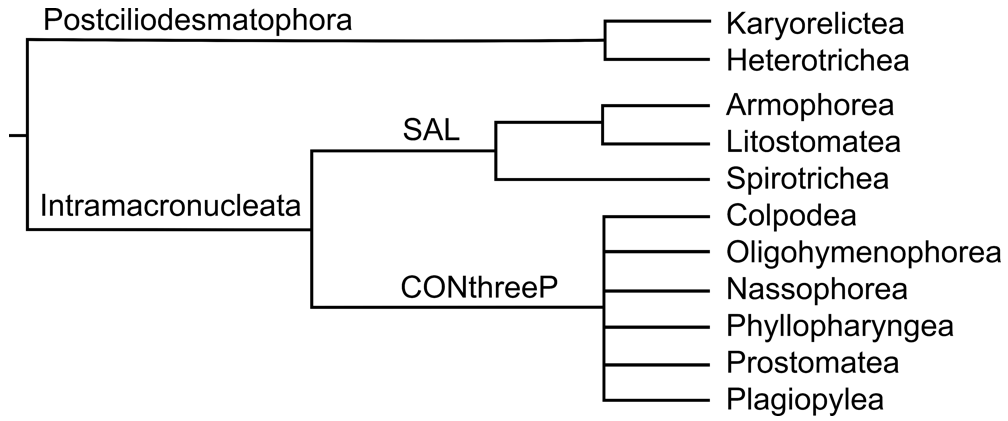
For taxonomy, we used Adl et al. system from 2019 as this system is comprehensive, made by specialists, and often followed in scientific papers. But not all classes or groups in this system are monophyletic in the ciliate SSU-rRNA phylogeny. Therefore, we forced monophyly of all ciliate classes based on this taxonomical system. It contains 11 ciliate classes that are grouped into several bigger clades, and those are finally grouped into these two primary ciliate clades.
Additionally, we included the recently established ciliate classes (Odontostomatea, Muranotrichea, and Parablepharismea) that are not in the Adl et al. system. All of these classes are in the vicinity of the class Armophorea, and all of them are monophyletic. One can decide how to treat these clades - separately or as one clade within the class Armophorea.
Lastly, there are also several ciliate taxa with an uncertain phylogenetic position. We treated them as in the Adl et al. 2019 system except for mesodinium species that we forced to the class Litostomatea, as the close relationship between mesodiniids and litostomateans was recently supported by phylogenomic data.